


|
핵심 제품 대부분 Class I 등록…“기술적 괴리 크다”
로킷헬스케어 미국 법인 로킷아메리카는 지난 4월 미국 증권거래위원회(SEC)에 제출한 나스닥 상장 증권신고서(S-1)에서 “3차원(3D) 바이오프린터, AI 기반 환부 모델링 소프트웨어, 일회용 재생 키트로 구성된 장기재생 플랫폼(ORP)를 통해 환자 맞춤형 재생 패치를 단일 스캔만으로 제작할 수 있다”고 투자자들에게 설명했다.
로킷헬스케어 기업설명(IR) 자료 역시 △세계 최초 의료용 3D 바이오프린터 △세계 최초 AI 환부 초정밀 모델링 앱(APP) △바이오잉크 제작 키트 등을 핵심 기술로 제시하며 '세계 최초 AI 초개인화 장기재생플랫폼 상업화 완료'를 강조하고 있다.
로킷헬스케어 측은 3D 바이오프린터에 대해 "독자적인 재생 니치(Niche) 작용 기전 및 기술을 기반으로 최적의 세포 재생 환경을 구축하고 상처 재발을 방지한다"고 설명했다. AI 앱은 "세계 최초 AI 환부 자동 모델링 기술과 자체 데이터 기반 알고리즘을 적용해 패치 정확도를 높였다"고 소개했다. 바이오잉크 제작 키트에 대해서는 "환자 MAECM 필터링을 통해 재생 능력을 보유한 바이오잉크로 변환한다"고 홍보했다.
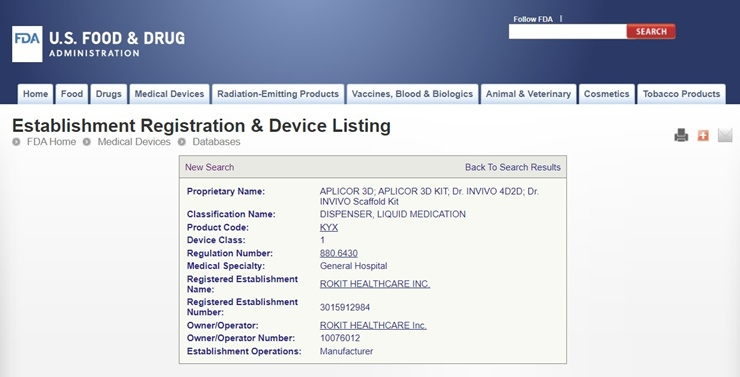
하지만 이데일리 제약·바이오 프리미엄 콘텐츠 팜이데일리가 FDA 의료기기 등록 데이터베이스를 직접 확인한 결과 실제 등록 내용은 회사 설명과 상당한 차이를 보였다.
3D 바이오프린터(Dr. INVIVO), 3D 바이오프린터 액세서리 키트(Dr. INVIVO Scaffold KIT), 인공지능(AI) 애플리케이션(AiD Regen)은 모두 Class I 으로 등록됐다. 3D 바이오프린터는 2020년 8월, AI 애플리케이션은 2021년 4월, 키트는 2022년 2월 각각 FDA 등록 절차를 거쳤다.
FDA 가이드라인에 따르면 의료기기는 위험도에 따라 Class I~III로 구분된다. 이 가운데 Class I은 가장 낮은 위험군으로 대부분 510(k) 사전심사 대상에서 면제된다. 주사기, 계량컵, 반창고 등이 대표적인 Class I 제품으로 알려졌다. 제조사는 FDA 등록 및 제품 목록 등재(listing) 절차를 거쳐 제품을 유통할 수 있다.
반면 Class II는 특별 규제(Special Controls)와 함께 통상 510(k) 허가 심사를 받아야 하며 Class III는 생명 유지 또는 중대한 위험 가능성이 있는 고위험 의료기기로 시판 전 승인(PMA)이 요구된다.
의료기기업계에서는 장기 재생 기술을 강조하는 플랫폼 핵심 제품들이 정작 Class I으로 등록된 것 자체가 회사 측 기술 설명과 규제 수준 사이 괴리를 보여준다고 지적한다.
의료기기 관련 바이오텍 고위 관계자는 "Class I은 인체 영향이 거의 없는 수준의 제한적인 사용 목적(Intended Use)으로 등록한 것"이라며 "Class I은 등록 서류만으로 가능하지만 실제 의료 현장에서 사용이 확대되려면 결국 클리니컬 베네핏(Clinical Benefit, 환자에게 실제로 유의미한 이득)을 입증해야 한다. 궁극적으로는 임상 데이터를 기반으로 Class II 이상 인허가가 필요하다"고 말했다.
이어 "재생 기능을 한다면 사용 목적 자체가 달라진다"며 "Class I이라는 것은 매우 낮은 수준의 사용 목적(Intended Use)으로 등록했다는 의미"라고 지적했다.
FDA 의료기기 등급은 제품의 물리적 형태 자체보다 제조사가 신고한 사용 목적에 따라 결정된다. 같은 장치라도 어떤 기능과 목적을 주장하느냐에 따라 적용 규제와 심사 수준은 크게 달라진다.
만약 로킷헬스케어가 3D 바이오프린터와 소모품 키트를 단순 기계 장치가 아니라 조직 재생 기능을 가진 의료기기로 FDA에 신고했다면 현재와 같은 Class I 심사 면제 범주에 포함되기 어려웠을 가능성이 높다. 재생 기능이 사용 목적에 포함될 경우 FDA는 통상 Class II 이상의 규제 체계를 적용하며 기존 유사 제품과의 실질적 동등성(510(k)) 또는 추가 안전성·유효성 자료 제출을 요구할 수 있다.
특히 기존 유사 제품이 없거나 고위험 기술로 판단될 경우에는 Class III 사전 시판 승인(PMA) 대상으로 분류될 가능성도 있다. 이 경우 임상시험을 포함한 안전성·유효성 검증 절차가 요구된다.
자가 지방세포 기반 바이오잉크 역시 FDA에 인체 세포·조직 기반 재생 제품(HCT/P)으로 신고할 경우 21 CFR 1271 규정 적용 대상이 될 수 있다. FDA는 최소 조작(Minimal Manipulation) 및 동종 사용(Homologous Use) 충족 여부를 검토하며, 이를 충족하지 못할 경우 보다 강화된 생물의약품 규제 체계 적용 가능성이 제기된다. 하지만 로킷헬스케어 측은 바이오잉크에 대해서도 재생 제품으로 신고하지 않았다.
|

FDA 등록 내용에 ‘재생 기능’ 표현 없어
실제 FDA 의료기기 등록 자료를 살펴보면 로킷헬스케어의 3D 바이오프린터(APLICOR 3D·Dr. INVIVO 4D2D)와 관련 키트(APLICOR 3D KIT·Dr. INVIVO Scaffold Kit)는 모두 액체 의약품 디스펜서(Dispenser, Liquid Medication) 제품군으로 등록돼 있었다. AI 앱 ‘AiD Regen’은 의료기기 데이터 저장 소프트웨어(Medical Device Data System·MDDS)로 분류됐다.
사용 목적 역시 재생 기술과는 거리가 있었다. FDA 규정(21 CFR 880. 6430)에 따르면 액체 의약품 디스펜서는 일정량의 액체 의약품을 분배하는 장치로 정의된다. AI 앱 역시 연결된 의료기기의 기능이나 설정값을 변경·제어하지 않으면서 의료기기 데이터를 전자적으로 전송·저장·변환하는 장치로 설명돼 있다. 쉽게 말해 의료기기 데이터를 저장·전달하는 기능 수준으로 실제 의료기기 작동 자체에는 관여하지 않는다는 의미다.
키트 제품의 경우 FDA UDI 데이터베이스(GUDID)상 '전자식 실험실 액체 분배 장치'(Laboratory liquid dispenser, electronic)로 기재돼 있었다. 사용 목적 역시 '저장소(병 또는 용기)에서 액체를 분배하도록 설계된 전자식 실험실 기기'로 설명됐다.
결국 핵심 제품들의 FDA 등록 내용 어디에도 장기 재생이나 조직 재생 기능은 포함되지 않은 셈이다. ORP를 구성하는 제품 가운데 유일하게 Class II로 분류돼 FDA 허가를 받은 제품은 지방분리기 아디나이저(Adinizer)였다. 다만 이 제품은 로킷헬스케어 자체 개발 제품이 아니라 BSL이라는 외부 업체가 개발한 제품이다.
FDA 설명에 따르면 아디나이저는 지방흡입 조직을 세척하고 혈액·오일을 제거한 뒤 일정 크기로 걸러주는 기능을 하는 미용 목적 제품으로 분류된다. 의료기기업계에서는 장기 재생 플랫폼 기업을 표방하는 로킷헬스케어 핵심 제품들이 대부분 Class I에 머물러 있고, FDA 등록 내용상 재생 기능 역시 확인되지 않는다는 점에서 기술적 우위나 혁신성을 설명하기 어렵다는 반응이 나온다.
실제 로킷헬스케어와 유사한 사업 모델을 가진 티앤알바이오팹은 3D 바이오프린팅, 세포외기질(ECM) 기반 바이오잉크, 폴리카프로락톤(PCL) 스캐폴드를 활용해 환자 맞춤형 조직재생용 인공지지체 사업을 진행 중이다. 이 회사 핵심 제품은 미국에서 Class II로 분류돼 약 3년간 임상 및 검증 절차를 거쳐 FDA 510(k) 허가를 진행하고 있다.
한 의료기기 바이오텍 대표는 "3D 바이오프린팅 기반 재생 의료기기를 판매하려면 결국 각국 규제기관 허가 절차를 거쳐야 한다. 의료기기 허가는 제품 등급에 따라 절차와 요구 데이터 수준이 완전히 달라진다"며 "3D 프린팅 기반 재생 기술은 쉽게 접근할 수 있는 영역이 아니고 대규모 임상과 수년간 검증 과정이 필요한 분야"라고 말했다.
팜이데일리는 로킷헬스케어 측에 미국에서 Class I로 분류된 장기재생플랫폼 핵심 제품 관련 질문을 했지만 답변을 피했다.


![김병주 ‘개인보증' 수용…홈플러스 운명, 다시 메리츠 손에[only 이데일리]](https://image.edaily.co.kr/images/Photo/files/NP/S/2026/07/PS26070300789t.1200x.0.jpg)



