식품의약품안전처는 최근 재심사 대상 의약품의 조사대상자의 수를 품목의 특성에 맞춰 산정하는 내용을 담은 ‘신약 등의 재심사 기준’ 일부 개정안을 행정예고했다. 이에 따라 제약사들은 신약과 개량신약에 한해 시판 후 의무적으로 진행해야 하는조사를 획일적으로 정하지 않고 품목 특성에 따라 유동적으로 정할 수 있게 했다. [단독]말많은 의약품 부작용 보고 제도 뜯어 고친다
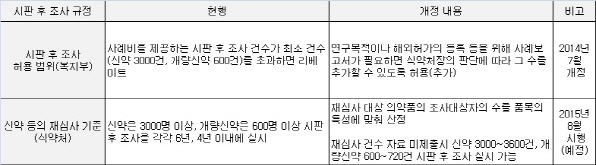
시판 후 조사(PMS)란 재심사 대상으로 지정된 신약·개량신약을 발매한 제약사가 일정기간 동안 정해진 건수의 부작용을 조사하는 제도를 말한다. 식약처는 신약은 발매 후 6년내 3000명 이상, 개량신약은 4년내 600명 이상을 대상으로 부작용 조사를 실시하도록 규정했다.
하지만 지난 2010년말 보건복지부는 약사법시행규칙을 개정하면서 신약 3000명, 개량신약 600명을 초과하는 부작용 조사에 참여하는 의사에게 사례비를 지급하면 리베이트로 처벌한다는 내용을 반영했다.
시판 후 조사는 제약사가 의사들에게 일정 금액을 지불하고 조사를 의뢰하는 방식으로 진행되는데, 제약사들이 처방 대가의 목적으로 이 제도를 리베이트 수단으로 악용하는 사건이 빈번해지면서 규제를 강화한 것이다. 가급적 많은 부작용 조사를 권고하는 식약처의 규정과는 상반된 내용이다.
복지부와 식약처의 규정이 부딪히면서 신약의 시판 후 조사는 부작용 점검 대상 3000명을 못 채워도 처분을 받고, 3000명을 초과해도 리베이트로 행정처분을 받는 불합리한 제도로 전락했다
실제로 한미약품(128940)의 ‘아모잘탄’, SK케미칼(006120)의 ‘엠빅스S’ 등이 부작용 조사를 많이 했다는 이유로 행정처분을 받았고 상당수 신약·개량신약 제품들이 행정처분 대상으로 지목받으면서 업계에서는 혼선이 가중됐다.
업계의 반발이 빗발치자 복지부는 지난해 약사법시행규칙을 개정하며 제도 개선에 나섰다. 의약품의 시판 후 조사와 관련 ‘연구목적이나 해외허가의 등록 등을 위해 사례보고서가 필요하면 식약처장의 판단에 따라 그 수를 추가할 수 있도록 한다’는 내용을 추가했다. 신약 3000건·개량신약 600건을 초과하면 무조건 리베이트로 처벌한다는 기존 입장에서 한발 물러선 셈이다.
여기에 식약처가 재심사 건수를 등 품목의 특성을 고려해 산출·결정할 수 있도록 결정하면서 제약사들은 부작용 조사에 따른 리베이트 행정처분 위험에서 5년만에 벗어나게 됐다.
제약사가 신약 등의 허가를 신청할 때 환자의 유병률과 같은 타당한 근거를 제출하면 식약처의 심사를 거쳐 재심사 조사 대상자를 조절할 수 있게 된다.
다만 신약과 개량신약 허가를 신청하는 제약사가 재심사 건수에 대한 자료를 제출하지 않을 경우 종전처럼 신약 3000건 이상, 개량신약 600건 이상을 의무적으로 부작용 조사를 실시해야 한다. 이때 최소건수의 20%(신약 600건, 개량신약 120건)까지는 별도로 변경신청하지 않아도 부작용 조사를 진행할 수 있다.
제약업계 관계자는 “해외에 수출하는 의약품의 경우 현지에서 가급적 많은 부작용 조사 자료를 요구하는 경우가 많다”면서 “이번 규제 완화로 연구활동이나 수출에도 숨통이 트일 것으로 기대된다”고 말했다.
|



!['36.8억' 박재범이 부모님과 사는 강남 아파트는[누구집]](https://image.edaily.co.kr/images/vision/files/NP/S/2026/02/PS26021500062t.jpg)
